
NanoBioSciences Group, Institute for Physical and Theoretical Chemistry, Technische Universität Braunschweig, Hans-Sommer-Strasse 10, 38106 Braunschweig
Small holes in aluminum films, called zeromode waveguides (ZMWs), can serve as ultra-small observation volumes for single-molecule spectroscopy at high, biologically relevant concentrations and are commercially used for real-time DNA sequencing [1]. To benefit from the single-molecule approach, each ZMW should be filled with one target molecule which is not possible with stochastic immobilization schemes by adapting the concentration and incubation time. We present DNA origami nano-adapters that by size exclusion allow placing of exactly one molecule per ZMW (Fig. 1b). The DNA origami nano-adapters thus overcome Poissonian statistics of molecule positioning and furthermore improve the photophysical homogeneity of the immobilized fluorescent dyes [2].
Funding by a starting grant (SiMBA, EU 261162) of the European Research Council (ERC), and the Volkswagen Foundation (86415 - 86416) is gratefully acknowledged.
[1] Eid J et al., Science 323, 133 (2009).
[2] Heucke S, Baumann F, Acuna GP, Severin PM, Stahl SW, Strackharn M, et al., Nano Lett. 14, 391 ( 2014).
Photonics and Optoelectronics Group, Ludwig-Maximilians-Universität München, Amalienstr. 54,80799 Munich (Germany)
Nanosystems Initiative Munich (NIM), Munich, Germany
DNA-directed self-assembly is one of the most powerful and versatile strategies to controllably organize colloidal nanoparticles using DNA as a surface ligand. Thereby assembly can be achieved by either DNA hybridization between two nanoparticles [1] or by using a three dimensional DNA template folded by the DNA origami technique.[2,3] Whereas the use of DNA objects enables the precise positioning of nanoparticles, site-selective alignment of anisotropic particles like gold nanorods (Au NRs) remains challenging. Au NRs are appealing building-blocks for the DNA-directed self-assembly of hierarchical heterostructures, especially due to their unique plasmonic properties. [4]
We have developed a robust strategy to functionalize Au NRs with a controlled number of single-stranded DNA molecules. We maintain colloidal stability throughout the process and enable at the same time cetyltrimethylammonium bromide (CTAB) removal.[5] The DNA-modified Au NRs are highly stable in biological buffers of high ionic strengths. Here, it will be shown how such NRs can be made active for hybridization with other DNA-functionalized nanoparticles at selective surface sites. Such site-selective DNA hybridization is important for the controlled bottom-up design of heterostructures with hierarchical order.
[1] J. Lee, G. Kim, J. Nam, J. Am. Chem. Soc. 134, (2012) 5456-5459.
[2] R. Schreiber, J. Do, E. Roller, T. Zhang, V. Schuller, P. Nickels, J. Feldmann and T. Liedl, Nature Nanotechol. 9 (2014), 74-78.
[3] R. Schreiber, N. Luong, Z. Fan, A. Kuzyk, P. Nickels, T. Zhang, D. Smith, B. Yurke, W. Kuang, A. Govorov and T. Liedl, Nat. Commun. 4 (2013) 1-6.
[4] C. Sönnichsen, T. Franzl, T. Wilk, G. von Plessen, J. Feldmann, O. Wilson, and P. Mulvaney, Phys. Rev. Lett. 88 (2002) 077402.
[5] B. Thierry, J. Ng, T. Krieg and H. Griesser., Chem. Comm. (2009), 1724-1726.
1 Leibniz Institute of Photonic Technology (IPHT), Albert-Einstein-Straße 9, 07745 Jena, Germany
2 Carl-Zeiss-Gymnasium Jena, Erich-Kuithan-Straße 7, 07743 Jena, Germany
The detection and therefore the amplification of DNA is an important process in various fields of science. The established technique for amplifying DNA, namely the PCR, is one of the most used reactions in the molecular biology. In 2000 a new procedure was published: the Loop-mediated isothermal amplification (LAMP) [1]. Because this technique is isothermal it can potentially be used as point of care detection more easily than PCR.
In the scope of a high school project we have developed a prototype of a device that is able to keep the probes at the needed temperature and allows an optical detection of the results afterwards. Our aim was to keep the production costs as low as possible. Therefore we mainly used well accessible and low-priced materials. To retain preferably user-friendly as well as the thermal system and the optical recognition is controlled by a common Android-based smartphone. The detection uses the camera of the smartphone.

- Fig. 1: Heating of the probes using common household halogen lamps
- Fig. 2: Optical detection by using an Android-based smartphone
- Fig. 2: Optical detection by using an Android-based smartphone
[1] Notomi T, et al 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Research 28:E63
Center for DNA Nanotechnology (CDNA), iNANO and Department of Chemistry, Aarhus University, 8000 Aarhus C, Denmark.
The idea behind our research is to use DNA as a programmable tool for directing the self-assembly of molecules and materials. The unique specificity of DNA interactions and our ability to synthesize artificial functionalized DNA sequences makes it the ideal material for controlling self-assembly and chemical reactions of components attached to DNA sequences. We have applied these concepts to assemble and covalently couple conjugated organic molecules1 and dendrimers.2 Recently, we extended this to DNA templated conjugation of DNA to proteins. In our studies of DNA origami we developed a method to image chemical reactions with single molecule resolution3 and to make a 3D DNA origami box with a controllable lid.4 More recently, we prepared a DNA-phenylene vinylene polymer and assembled it on DNA origami for studies of electronic and optical properties. In extension of this a method for self-assembly of DNA origami and single stranded tile structures at room temperature will also be presented.5

- Fig. 1: Schematic illustration and AFM image of poly(DNA-phenylene vinylene) on DNA origami.
1. Ravnsbæk; J. B et al. Angew. Chem. Int. Ed. 2011, 50, 10851–10854.
2. Liu, H. et al. J. Am. Chem. Soc. 2010, 132, 18054-18056.
3. Voigt, N. V. et al. Nature Nanotech. 2010, 5, 200-205.
4. Andersen, E. S. et al. Nature 2009, 459, 73-76.
5. Zhang, Z. et al. Angew. Chem. Int. Ed. 2013, 52, 9219
a Medway School of Pharmacy, University of Kent, Chatham Maritime, Kent, United Kingdom
b Commonwealth Scientific and Industrial Research Organisation, Materials Science and Engineering, Australia
c Department of Chemistry and Biochemistry, University of California, Santa Barbara, USA
Nucleic acid diagnostics, often referred to as ‘molecular diagnostics’ (in seeming oblivion to the molecular nature of proteins and metabolites), measure DNA or various types of RNA in order to assay particular genomic or genetic details of a patient, or to assay nucleic acid sequences unique to invading pathogens. In POC diagnostics devices, sample volumes are often measured in microliters, and little or no user manipulation should be necessary. Key issues are the mode of immobilization of the capture DNA probes at the appropriate surface density, reactivity in analyte-detection molecule capture analyses, and the contribution of non-specific binding.
Here we present a very interesting concept for reagent free, nucleic acid assay test consisting of a panel of 2 miRNAs that act as a breast cancer-specific diagnostic for early detection of disease and monitoring treatment response. The approach is based on combination of engineering of the physico-chemical properties of the substrate surface and the capture probe orientation changes. We will demonstrate that the capture probe immobilized on suitable surface can selectively hybridize to its complementary target. The resulting double helix changes its orientation to vertical position thus enabling the detection based on fluorescence quenching phenomenon (Figure 1). Interestingly, such orientation changes are very sensitive to the target sequence, allowing to specifically discriminate between the two studied miRNAs that differ by only three bases [1]. To study the consequences of the key surface properties on the orientation and the activity of surface-bound DNA probes on different types of surfaces, we employed a label-free method called total internal reflection ellipsometry (TIRE) and a Dual Polarization Interferometry (DPi). Comprehensive data on characterization of the thin-films using other supporting techniques such as Atomic Force Microscopy, X-ray Reflectivity, water contact angle and Total Internal Reflection Fluorescence microscopy will be presented as well.
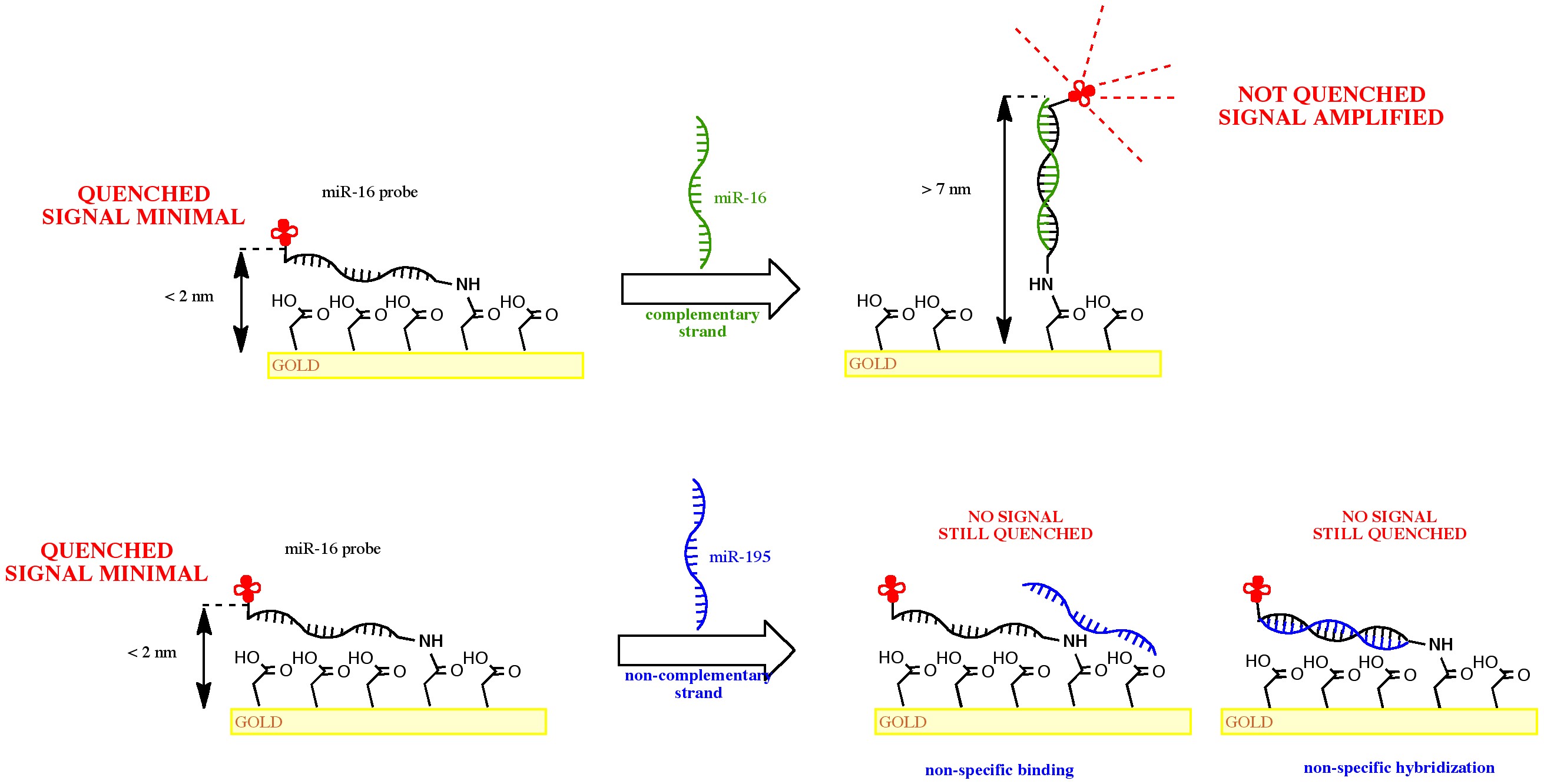
- Fig. 1: The reagent-free detection concept based on fluorescence quenching phenomenon specific to probe –double helix orientation changes
[1] App. Mater. Interfaces, 2011, 3 (12), 4640–4648
B CUBE - Center for Molecular Bioengineering, Arnoldstraße 18, 01307 Dresden
Magnetic tweezers[1] allow the manipulation of individual DNA molecules through external force and torque. Torque is stored in DNA predominantly in two ways: as twist of the individual single DNA strands around each other and as writhe, when plectonemic loops are formed along the molecule’s extension.
When adding additional turns to DNA molecules, two torque regimes exist, separated by the onset of the first plectonemic loop. The transition between both regimes is depicted in Fig. 1 as a change from the central to the flanking parts. Brutzer et al. [2] have studied the torque-dependent DNA molecular energetics at positive superhelicity. Recently, Salerno et al. [3] have shown that - at forces exceeding a critical force - negative superhelicity is accompanied by DNA double strand melting and therefore increased fluctuations of the molecular extension (see Fig. 2). In other magnetic tweezers experiments, the onset of DNA denaturation and formation of secondary structures as an extrusion of cruciform hairpin structures was shown to lead to large changes in DNA extension[4].
In our force-dependent DNA supercoiling experiments, we investigate whether local secondary structure formation is a generally occurring phenomenon in the breakdown of the linear torque storage regime at negative superhelicity and in critical force conditions. Screening our sample sequence for possible hairpin structures and imperfect inverted repeats, we investigate whether DNA secondary structure formation can be observed in single-molecule magnetic tweezers experiments of DNA double strand melting.
Funding by the BMBF is gratefully acknowledged.

- Fig. 1:Histogram-plot of DNA supercoiling at low-force conditions (F= ???).
Fig. 2: Histogram-plot of DNA supercoiling with increased fluctuations at negative turns at higher-force conditions (F = ???).
[1] Z. Bryant, F. C. Oberstrass, and A. Basu, Current Opinion in Structural Biology 22, 304 (2012).
[2] H. Brutzer, N. Luzzietti, D. Klaue, and R. Seidel, Biophys. J. 98, 1267 (2010).
[3] D. Salerno, A. Tempestini, I. Mai, D. Brogioli, R. Ziano, V. Cassina, and F. Mantegazza, Phys. Rev. Lett. 109, 118303 (2012).
[4] T. Ramreddy, R. Sachidanandam, and T. R. Strick, Nucleic Acids Research 39, 4275 (2011).
*Department of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China
DNA phosphorylation is an important regulatory process in a variety of cellular processes, such as nucleic acid metabolism and DNA damage repair.[1] Polynucleotide kinase (PNK) catalyzes the phosphorylation of oligonucleotides at the 5′-hydroxyl end, and the catalytic activity of PNK is critical for repairing DNA strand breaks induced by endogenous or exogenous agents.[2] Therefore, the accurate detection of PNK activity is of considerable importance in biochemical and molecular biology studies. In this study, a label-free, oligonucleotide-based, switch-on luminescence detection method for T4 polynucleotide kinase activity has been developed using a novel G-quadruplex-selective luminescent Ir(III) complex probe (Fig. 2). The principle of the assay is depicted in Fig. 1, the hairpin oligonucleotide HG55 consists of a 3′-G-quadruplex-forming sequence (depicted green), its complementary 5′-sequence (depicted blue) and a short linker region (depicted red). 5′-Phosphorylation of HG55 by T4 PNK coupled with digestion of the 5′-strand by λ exo liberates the G-quadruplex motif, which is detected by 1 with a switch-on luminescent response (Fig. 3). The application of the assay for screening potential T4 PNK inhibitors is also demonstrated. To our knowledge, this is the first metal-based assay for PNK activity.

- Fig. 1: Schematic representation of the G-quadruplex-based luminescence switch-on detection strategy for T4 PNK activity using the G-quadruplex-selective Ir(III) complex 1.
Fig. 2: Chemical structure of cyclometallated Ir(III) complex 1.
Fig. 3: Luminescence spectra of the 1/HG55 system in response to various concentrations of T4 PNK.
[1] C. J. Whitehouse et al., Cell 104 (2001) 107-117.
[2] L. K. Wang et al., EMBO J 21(23002) 3873-3880.
A Synergy for Top-Down and Bottom-Up Nanofabrication
University of California San Diego, Department of Nanoengineering and Department of Bioengineering, La Jolla, CA, USA 92093-0448, , mheller@ucsd.edu, 760-415-1962
Intrinsic programmability and other unique properties of DNA allow it to be used as a self-assembly nanostructure material, easily functionalized with nanoparticles and other entities, and as a UV sensitive write material for patterning. Thus, DNA has shown considerable potential for both top-down photolithographic and bottom-up self-assembly nanofabrication. However, using present methods for UV patterning on DNA substrates limits the hybridization of complementary DNA sequences to only the DNA in the patterned areas that was not exposed to UV. Such UV single-write methods greatly restrict the full potential of DNA for further programmed self-assembly after patterning. We have now been able to demonstrate a DNA double-write process, which overcomes this limitation. Using specially designed DNA constructs immobilized on a glass substrate, UV patterning results in two distinct binding identities to which two different complementary DNA sequences can be hybridized. This provides a major advantage that allows DNA based self-assembly as well as further UV patterning to be carried out in both the UV exposed and non-exposed areas. Using the DNA double write process, UV patterning has been carried out with 500nm resolution.
Michael J. Heller received his Ph.D. in Biochemistry from Colorado State University in 1973. He was an NIH Postdoctoral Fellow at Northwestern University from 1973 to 1976. Dr. Heller was supervisor of the DNA Technology Group at Amoco Corporation from 1976 to 1984. During that time he carried out bioengineering and recombinant DNA engineering work on plants, algae and photosynthetic bacteria for energy and chemical production, and oversaw the companies sponsored research at the Cetus Corporation. Dr Heller was Director of Molecular Biology at Molecular Biosystems from 1984 to 1987. He was a co-founder of Integrated DNA Technologies and served as President and Chief Operating Officer from 1987 to 1989. Dr Heller was a co-found of Nanotronics and Nanogen and served as the Chief Technical Officer from 1993 to 2001. Nanogen carried out the successful development of microelectronic DNA array technology for clinical genotyping applications. Dr. Heller is now a Professor in the Departments of Nanoengineering and Bioengineering at the University California San Diego. He has also recently co-founded a new company called Biological Dynamics (started by his PhD student) which is developing new cancer diagnostics technology. Dr. Heller has extensive industrial experience in biotechnology, biomedical and clinical diagnostic devices and nanotechnology; with particular expertise in the areas of DNA probe diagnostics, DNA synthesis, fluorescent-based detection technologies and electric field assisted self-assembly of DNA nanocomponents. Dr. Heller has a respectable publication record, has over 45 issued US Patents and has been an invited speaker to a large number of scientific conferences and meeting. Dr. Heller has been a panel member for the White House (OSTP) National Nanotechnology Initiative 1999/2000; the NAS (NAE) Review of National Nanotechnology Initiative 2001-2002; the NAS(NAE) – Engineer for the 2020 - 2001/2002; and has also been involved in a number of NSF Nanotechnology Workshops.
A Synergy for Top-Down and Bottom-Up Nanofabrication
University of California San Diego, Department of Nanoengineering and Department of Bioengineering, La Jolla, CA, USA 92093-0448, , mheller@ucsd.edu, 760-415-1962
We present and compare three optical methods based on spectroscopy of noble metal nanoparticles exhibiting Localized Surface Plasmon Resonances (LSPR) for their application as label free optical bio-sensors.
By an external incident light beam, density oscillations of the nanoparticle’s conducting electrons are induced at a specific frequency, which is known as Localized Surface Plasmon Resonance (LSPR). This occurring resonance band is sensitive to changes of the surrounding medium, which gives the opportunity to utilize them as label-free bioanalytical sensors. Biomolecules can be bound directly on the nanoparticle’s surface, which leads to a change of the local refractive index and a shift of the peak wavelength.
We present the detection of binding events on nanoparticle layers and on single nanoparticles. The measurements of nanoparticles layers were performed in real time and showed strong LSPR signal-to-noise signal ratio, but decreased spatial resolution and no multiplexing. The real-time single LSPR sensing on the other hand allows detection of adsorption of a small number of molecules with a high spatial resolution, but it is limited to simultaneous detection only of one nanoparticle in our standard experimental set-up, which is a combination of a microscope and dispersion based spectrometer. To overcome these limitations a third home-built set-up will be introduced, which is a combination of dark-field microscopy and Fourier Imaging Spectroscopy. It allows signal collection of more than 50 independent nanoparticles in parallel.
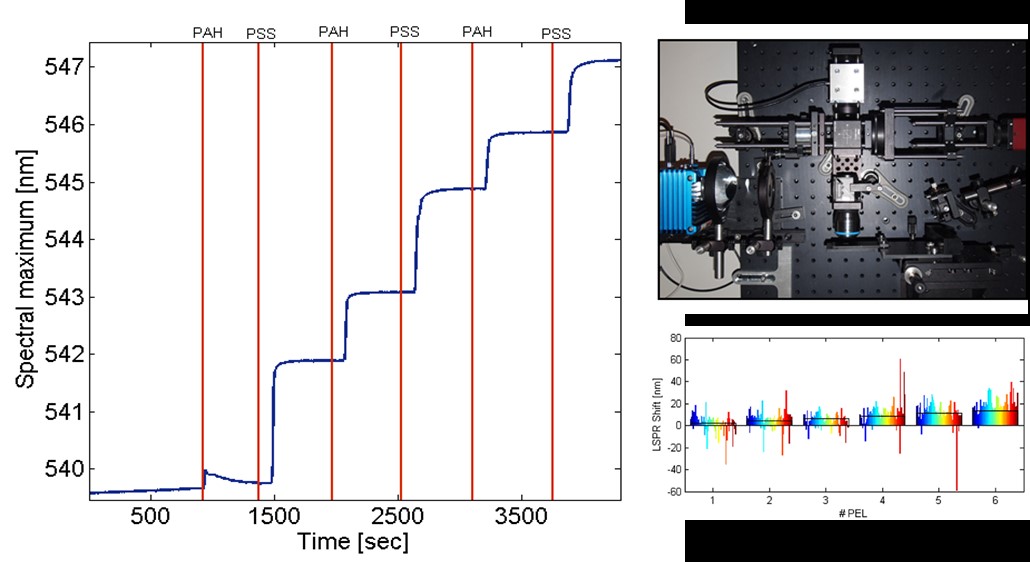
- Fig. 1: Left – Real time observation of nm thin polyelectrolyte layer deposition on densely immobilized nanoparticles. Top right – A picture of the setup of the Fourier transform imaging spectrometer. Bottom right – Spectral shift of localized surface plasmon resonance of 72 single gold particles in dependence of the number of deposited polyelectrolyte layers (PEL). Every color bar corresponds to the shift of one single nanoparticle and the black boxes represent the predicted theoretical shifts for ideally deposited polyelectrolyte layers.
Leibniz Institute of Photonic Technology, Albert-Einstein-Straße 9, 07745 Jena, Germany
DNA origami technique is a powerful tool for the precise positioning of plasmonic nanostructures in the range of a few nanometers. DNA-nanoparticle structures are usually produced in two steps divided in the folding of the DNA origami and a subsequent conjugation with functionalized nanoparticles. A simultaneous assembly could not only increase the yield but also allows the realization of more complex plasmonic constructs, especially for a three dimensional shape. Such a one-pot solution demands an optimization of the folding protocols.
Our investigations of the DNA folding process without denaturing conditions are also discussed like a self-assembly of DNA origami even at room temperature. This would be also promising for a one-pot folding of DNA and thermo-sensitive biomolecules like enzymes.
For DNA-nanoparticle structures, we present a method where the nanoparticles are functionalized with staple strands directly involved in the folding process and therefore supported the one-pot self-assembly of DNA-nanoparticle structure.
The DNA surface can work as a nanobreadbord for spherical or anisotropic nanoparticles like silver nano-prisms. Latter ones are highly interesting because of their plasmonic properties but are challenging in their stability. Here the silver nano-prisms were covered with a very thin gold layer and conjugated with DNA to solve this problem.

- Fig. 1: Folding of DNA rectangles at constant temperature without a prior denaturation
- Fig. 2: self-assembly of DNA-nanoparticles structures in a one-pot solution
- Fig. 3: gold coated silver nano-prism and 5 nm gold nanoparticles
[1] P.W. Rothemund (2006). Nature, 440 (7082), 297–302.
[2] Ding et al.(2010). J Am Chem Soc, 132, 3248-3249
1Max Planck Institute for Intelligent Systems, Stuttgart, Germany
2Fakultät für Physik, Ludwig-Maximilians-Universität, München, Germany
3Department of Physics and Astronomy, Ohio University, Athens, Ohio 45701, USA
†Present address: Clarendon Laboratory, University of Oxford, UK
*E-mail: kuzyk@is.mpg.de
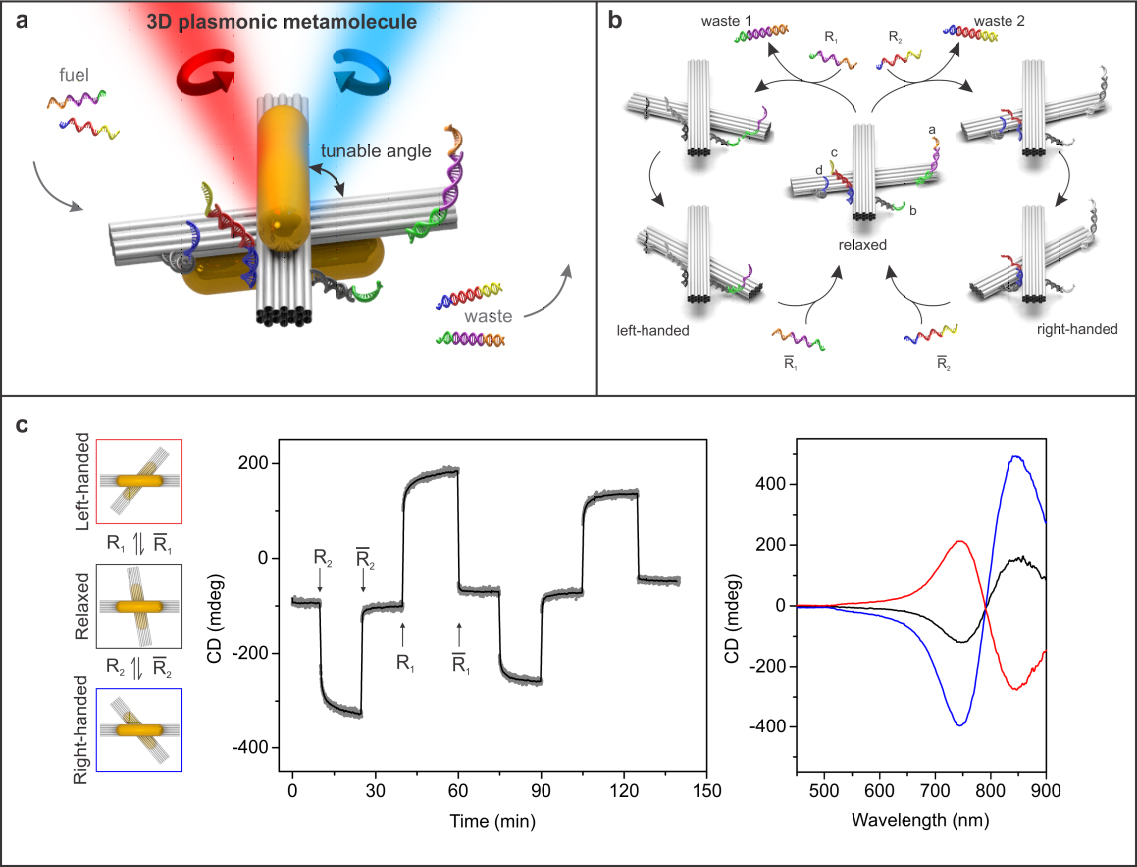
A reconfigurable plasmonic nanosystem combines an active plasmonic structure with a regulated physical or chemical control input. Such plasmonic devices hold great promise for applications in adaptable nanophotonic circuitry and optical molecular sensing. There have been considerable efforts on integration of plasmonic nanostructures with active platforms using top-down techniques. However, such plasmonic nanostructures are often restricted to two-dimensional substrates, showing desired optical response only along specific excitation directions. Also, realization of structural reconfigurability in the visible wavelength range remains challenging due to the static nature of top-down techniques. Here we lay out a multi-disciplinary strategy to create reconfigurable 3D plasmonic metamolecules, which execute DNA-regulated conformational changes on the nanoscale. In one role, DNA serves as construction material to organize plasmonic nanoparticles in 3D. In the other role, DNA is used as fuel for driving the metamolecules to distinct conformational states. Simultaneously, the 3D plasmonic metamolecules can work as optical reporters, which transduce their conformational information in situ into circular dichroism (CD) changes in the visible wavelength range.
1Fraunhofer Institute for Cell Therapy and Immunology (IZI), DNA Nanodevices unit, Leipzig, Germany
2University of Cologne, Department of Chemistry, Institute of Biochemistry, Cologne, Germany
3Friedrich Schiller University Jena, School of Medicine, Jena, Germany
DNA is a powerful building block which allows the programmed self-assembly of molecular scaffolds, cages and multifunctional carriers with nanoscale dimensions by the nature of predictable base pairing [1]. These structures can function as carriers to deliver functional biomolecules including proteins, antibodies or other therapeutic molecules to malignant or otherwise diseased cells.
Our early work uses anti-cancer drug paclitaxel (Taxol®). While primarily used as a microtubule-targeting drug, paclitaxel also associates non-covalently with double-stranded DNA and therefore can be attached to DNA origami-based carriers and delivered to cancer cells. Unlike intercalation-based drugs like doxorubicin which distort the three-dimensional form of origami-based carriers [2], paclitaxel binds to the groove of the double-helix of DNA without distorting its structure. For evaluating the efficacy of paclitaxel-loaded carriers, properties such as loading capacity, release rate and their effect on cell viability and mechanical properties are assessed.
vA small but very stable DNA nanostructure is the DNA-tetrahedron [3], which consists of four oligonucleotides which hybridize to form a wireframe tetrahedron with double-stranded edges of less than 10 nanometers. We want to use this structure to both gain greater understanding and modulate how DNA nanostructures enter cells by tracking the uptake dynamics and compartmentalization. Previous studies have shown that internalized DNA nanostructures were observed to co-localize with endosomal markers [4]. In order to access many cytosolic, nuclear or mitochondrial pathways which are potential targets for therapies, the ability to achieve a finer control compartmentalization is required. To study the dynamics of the cellular uptake of DNA-nanostructures, we attach short, mostly positively charged peptides derived from protein-transduction domains to the previous mentioned DNA tetrahedron. These so-called cell-penetrating peptides (CPPs) can be internalized in most cell types and, more importantly, allow the cellular delivery of conjugated or fused biomolecules [5]. By tracking the uptake dynamics and compartmentalization of fluorescently labeled tetrahedron-CPP-molecules, we hope to gain a greater control over how DNA-nanostructures enter cells, which is a fundamental question for their eventual application to regenerative medicine and cell therapy.
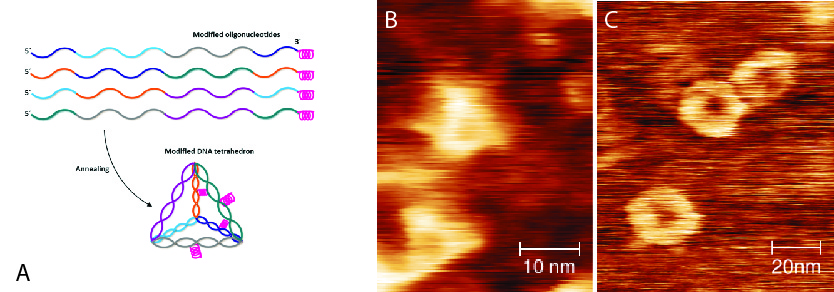
- Fig. 1: DNA tetrahedron.
A) schematic model of modified tetrahedron 3x20bp),
B) AFM image of unmodified tetrahedra,
C) AFM image of Cpp-linked tetrahedra.
[1] P.W. Rothemund (2006). Nature,440 (7082), 297–302.
[2] Y.-X. Zhao, A. Shaw, X. Zeng, E. Benson, A. M. Nyström, and B. Högberg (2012). ACS Nano, 6 (10), 8684-8691
[3] R.P. Goodman, I.A. Schaap, C.F. Tardin et al. (2005). Science, 310 (5754), 1661–1665.
[4] D. Smith, V. Schüller, C. Engst, J. Rädler& T. Liedl (2013). Nanomedicine, 8 (1), 105-121.
[5] J.P. Richard, K. Melikov, E. Vive´s, C. Ramos, B. Verbeure, M.J. Gait, L.V. Chernomordik, B. Lebleu (2003). J. Biol. Chem., 278, 585– 590.
1Fraunhofer Institute for Cell Therapy and Immunology (IZI), DNA Nanodevices unit, Leipzig, Germany
2Friedrich Schiller University Jena, School of Medicine, Jena, Germany
3University of Cologne, Department of Chemistry, Institute of Biochemistry, Cologne, Germany
The development and administration of hydrophobic drugs is often difficult due to their poor solubility in the bloodstream. To improve the delivery and bioavailability of certain drugs, nanoparticles which function as drug vehicles, are already used in medical therapeutics. DNA nanostructures are good carriers for naturally DNA-binding drugs (e.g., doxorubicin1, siRNA2), but they do not bind to hydrophobic drugs per se. In order to assemble both, amphiphilic polymers such as PEG are ideal intermediates with many benefits. The water-soluble PEG molecule can be covalently bound to several outward pointing staple strands, which are then folded to a desired DNA-origami, for example a six helix bundle (6HB). There it can function as a sponge for hydrophobic molecules around the origami structure.
The PEGylated and drug loaded DNA origami could improve the application of hydrophobic drugs in therapeutic medicine, and even provide a possible route by which drugs abandoned during the course of development due to solubility problems may be “revived”. Moreover, it is possible to add functional groups to the non-PEGylated part of the DNA origami.

- Fig. 1: Schematic demonstration of a 6HB loaded with hydrophobic drug molecules. A) 6HB (blue) partly covered with covalently-bound PEG (red); hydrophobic drugs (green), bind non-covalently to PEG. The longitudinal section shows functional parts of the origami, where e.g. antibodies (purple) can attach. B) Cross-section of modified 6HB.
[1] Zhao, Y., Shaw, A., Zeng, X. & Benson, E. DNA origami delivery system for cancer therapy with tunable release properties. ACS … (2012). at http://pubs.acs.org/doi/abs/10.1021/nn3022662
[2] Lee, H . et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 7, 389–93 (2012).
Chemical Nanoscience Laboratory, School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
Here we report the polymerization of pyrrole onto DNA origami ‘T’-nanostructures. The templating of -DNA with a range of conductive polymers has been explored and the conductivity of the 1D nanowires produced has been demonstrated. The fabrication of these 1D nanostructures is relatively straightforward but their integration into electronic circuits is more challenging. Circuit junctions and interconnects are required and recent work has shown that origami ‘T’ can be metalized, suggesting that DNA origami has a role to play in complex architectures Successful metallization of DNA origami with either gold or palladium was demonstrated and the results indicated that origami structures are sufficiently stable, do not undergo significant structural deformation during metallization. However, the formation of polymer templated DNA origami structures carries two further challenges that need to be addressed. Firstly; determination of the selectivity required to polymerize pyrrole on densely packed dsDNA within the relatively large origami structures (ca 60 nm × 80 nm compared to 2 nm × 17 m of -DNA), and secondly; integration of the modified origami structures with a substrate that is suited to electronic integration. This talk describes our efforts to fabricate DNA origami templated conducting polymers and deposit them on a silicon wafer in order to address these challenges.

- Fig. 1: Tapping mode AFM images. Left: DNA origami “T-shapes” before addition of pyrrole and Right:after treatment with pyrrole showing self-assembled conductive 2D-network formation.
Institute of Chemistry and Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, 91904 Israel
DNA and DNA-based polymers have been at the focus of molecular electronics owing to their programmable structural versatility. The variability in the measured molecules and experimental setups, caused largely by the contact problem, has produced a wide range of partial or seemingly contradictory results, highlighting the challenge to transport significant current through individual DNA-based molecules. A well-controlled experiment that would provide clear insight into the charge transport mechanism through a single long molecule deposited on a hard substrate has never been accomplished. I will report on detailed and reproducible charge transport in G4-DNA, adsorbed on a mica substrate. Using a novel benchmark process for testing molecular conductance in single polymer wires, we observed currents of tens to over 100 pA in many G4-DNA molecules over distances ranging from tens to over 100 nm, compatible with a long-range hopping between multi-tetrad segments. With this report, we answer a long-standing question about the ability of individual polymers to transport significant current over long distances when adsorbed a hard substrate, and its mechanism. These results may re-ignite the interest in DNA-based wires and devices towards practical implementation of these wires in programmable circuits
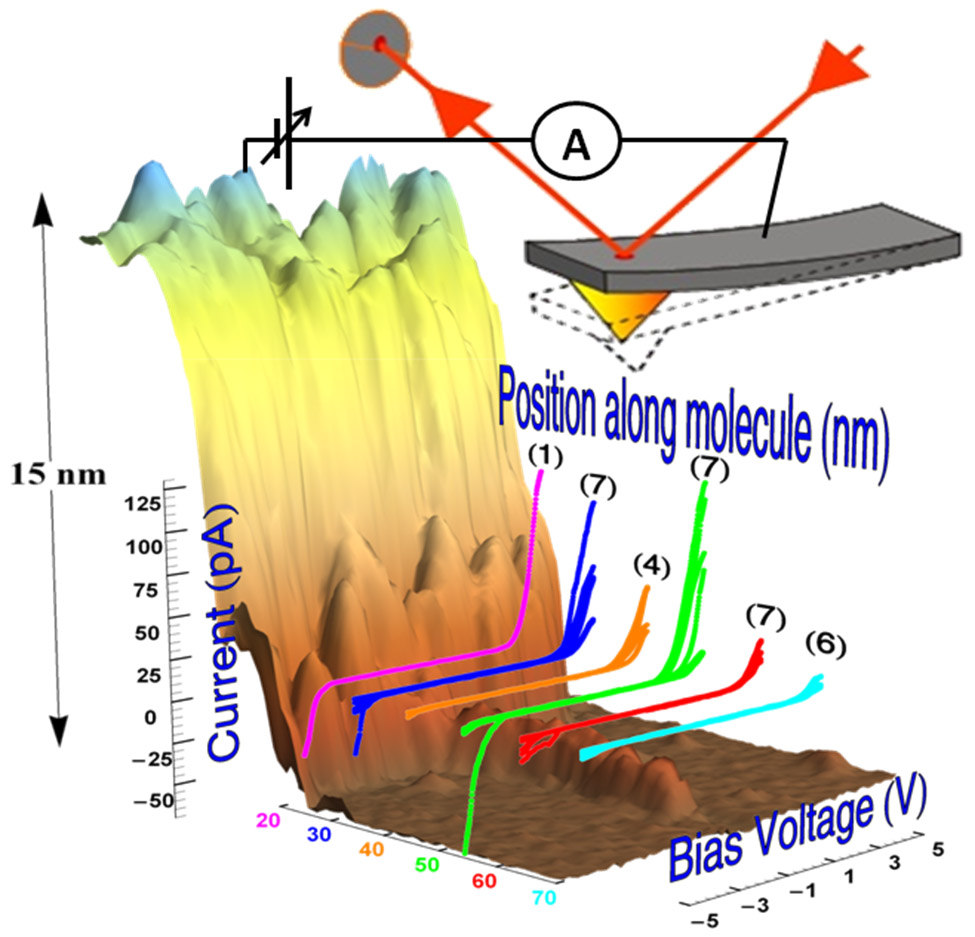
[1] "Direct measurement of electrical transport through DNA molecules", Danny Porath, Alexey Bezryadin,Simon de Vries and Cees Dekker, Nature 403, 635 (2000).
[2] "Charge Transport in DNA-based Devices", Danny Porath, Rosa Di Felice and Gianaurelio Cuniberti, Topics in Current Chemistry Vol. 237, pp. 183-228 Ed. Gary Shuster. Springer Verlag, 2004.
[3] “Direct Measurement of Electrical Transport Through Single DNA Molecules of Complex Sequence”, Hezy Cohen, Claude Nogues, Ron Naaman and Danny Porath, PNAS 102, 11589 (2005).
[4] “Long Monomolecular G4-DNA Nanowires”, Alexander Kotlyar, Nataly Borovok, Tatiana Molotsky, Hezy Cohen, Errez Shapir and Danny Porath, Advanced Materials 17, 1901 (2005).
[5] “Electrical characterization of self-assembled single- and double-stranded DNA monolayers using conductive AFM”, Hezy Cohen et al., Faraday Discussions 131, 367 (2006).
[6] “High-Resolution STM Imaging of Novel Poly(G)-Poly(C)DNA Molecules”, Errez Shapir, Hezy Cohen, Natalia Borovok, Alexander B. Kotlyar and Danny Porath, J. Phys. Chem. B 110, 4430 (2006).
[7] "Polarizability of G4-DNA Observed by Electrostatic Force Microscopy Measurements", Hezy Cohen et al., Nano Letters 7(4), 981 (2007).
[8] “Electronic structure of single DNA molecules resolved by transverse scanning tunneling spectroscopy”, Errez Shapir et al., Nature Materials 7, 68 (2008).
[9] “A DNA sequence scanned”, Danny Porath, Nature Nanotechnology 4, 476 (2009).
[10] “The Electronic Structure of G4-DNA by Scanning Tunneling Spectroscopy”, Errez Shapir, et.al., J. Phys. Chem. C 114, 22079 (2010).
[11] “Energy gap reduction in DNA by complexation with metal ions”, Errez Shapir, G. Brancolini, Tatiana Molotsky, Alexander B. Kotlyar, Rosa Di Felice, and Danny Porath, Advanced Maerials 23, 4290 (2011).
[12] "Quasi 3D imaging of DNA-gold nanoparticle tetrahedral structures", Avigail Stern, Dvir Rotem, Inna Popov and Danny Porath, J. Phys. Cond. Mat. 24, 164203 (2012).
[13] "Comparative electrostatic force microscopy of tetra- and intra-molecular G4-DNA", Gideon I. Livshits, Jamal Ghabboun, Natalia Borovok, Alexander B. Kotlyar, Danny Porath, submitted (2014).
[14] "Long-range charge transport in single G4-DNA molecules", Gideon I. Livshits et. al., submitted (2014).
Physical and Theoretical Chemistry - NanoBioScience, TU Braunschweig, Hans-Sommer-Strasse 10, 38106 Braunschweig, Germany
* Centro de Investigaciones en Bionanociencias (CIBION), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Godoy Cruz 2390, C1425FQD Buenos Aires, Argentina
DNA origami is a powerful tool for the construction of hybrid nanosize functional objects with a very high precision [1]. Here we demonstrate nanoantennas consisting of three dimensional DNA origami and gold nanoparticles [2]. These nanoparticles (forming monomers or dimers) exhibit a hotspot where an organic dye is positioned. At a 17 nm gap between two 80 nm gold nanoparticles, focusing light at the nano-scale led to a fluorescence enhancement of 300 fold. In addition, we show the increase of photostability in the vicinity of gold nanoparticles [3] and dependency with nanoparticle size. The results are rationalized with the aid of numerical simulations using FDTD.
Funding by a starting grant (SiMBA, EU 261162) of the European Research Council
(ERC).is gratefully acknowledged.
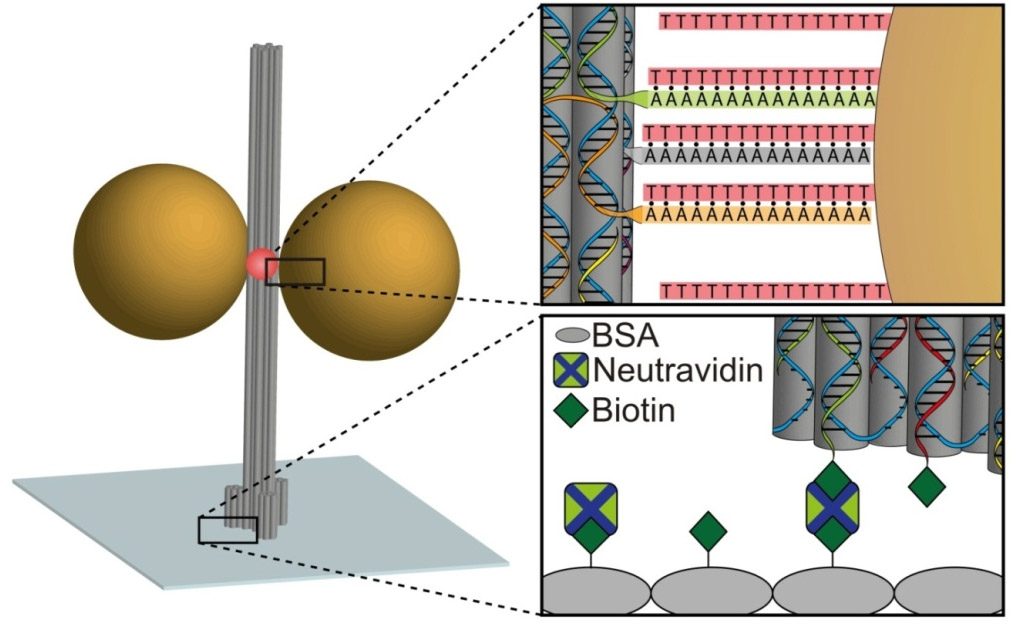
- Fig. 1: Sketch of the DNA origami pillar labeled with organic dye (red) immobilized on a glass surface. Gold nanoparticles of different sizes can bind, via a set of oligonucleotide chains laterally.
[1] P. W. Rothemund, Nature 440, 297 (2006).
[2] F. Möller, P. Holzmeister, T. Sen, G. P. Acuna and P. Tinnefeld, Nanophotonics 2, 167 (2013).
[3] J. Pellegrotti, G. P. Acuna, A. Puchkova, P. Holzmeister, A. Gietl, B. Lalkens, F. D. Stefani and P. Tinnefeld, Nature Communication, submitted 2014
Fraunhofer Institute for Cell Therapy and Immunlogy IZI, Perlickstraße 1, 04103 Leipzig, Germany.
Brick-based DNA nanostructures (Fig. 1) are a powerful approach for designing and creating DNA nanostructures at high speed [1]. As its name implies, the structures are constructed from a collection of several hundred synthetic single-stranded DNA oligonucleotide “bricks”, designed to link to neighboring bricks through complementary base pairing. From a predefined pool made up of several thousand DNA strands which collectively form a 3D canvas (Fig. 2, Fig. 3), nearly any arbitrary structure can be created in a matter of days. Due to its modularity and ability to be integrated with computer-assisted automated design and production, this technique is promising for potential industrial-scale applications. However, this necessitates an intimate understanding of the molecular assembly process so that factors such as production yield, speed of assembly and basic design can be fully optimized.
Studies on DNA origami have shown that scaffolded nanostructures can assemble within a relatively narrow temperature range [2], possibly due to cooperative effects arising from the long underlying scaffold strand. While brick-based DNA nanostructures are lacking this underlying scaffold, the highly regular pattern of hybridization linking together neighboring bricks could also give clues into optimization strategies for the assembly process. First estimations on brick-based DNA nanostructures indicated that during the thermal-based assembly, initial outer “shell” sections could be formed at higher temperatures (Fig. 4) followed by the hybridization of the inner portions of the structures at much lower temperatures. More exact nearest-neighbor thermodynamic [3] calculations of melting temperatures for hybridized sections of neighboring bricks pointed out two broad temperature ranges. The higher of these temperature ranges may form outer-shell sections which could act as nuclei for successive assembling. Real-time fluorometric assembling experiments will display the relevance of this theory. Preliminary experimental data show some similarity with previously described calculations of melting temperatures. Future experiments are planned to determine the influence of outer-shell sections in the entire assembling process.
Funding by Fraunhofer-Attract program is gratefully acknowledged.

-
Fig. 1: TEM-image of full block
Fig. 2: Voxel Simulation of 3D canvas.
Fig. 3: Strand simulation of 3D canvas
Fig. 4: Strand Simulation of outer “shell” sections
[1] Ke Y., Ong L.L., Shih W.M. & Yin P. (2012) Three-Dimensional Structures Self-Assembled from DNA Bricks. Science, 338: 1177-1183.
[2] Sobczak J.J.-P., Martin Th.G., Gerling Th. & Dietz H. (2012) Science, 338: 1458-1461. Wei X., Nangreave J., Jiang S., Yan H. & Liu Y. (2013). JACS, 135: 6165-6176.
[3] SantaLucia J. Jr. (1998) PNAS, 95: 1460-1465. Ahsen N. v., Wittwer C.T. & Schütz E. (2001).. Clinical Chemistry, 47,11: 1956-1961.
1) Cluster of Excellence Center for Advancing Electronics Dresden/ TU Dresden, Germany (e-mail : thorsten-lars.schmidt@tu-dresden.de);
2) Cluster of Excellence Macromolecular Complexes/ Goethe University Frankfurt am Main, Germany;
3) Dana-Farber Cancer Institute/ Department of Biological Chemistry and Molecular Pharmacology,/ Wyss Institute for Biologically Inspired Engineering at Harvard, Boston, MA, USA.
Dervan-Type Polyamides are a class of heteroaromatic polymers that can be programmed to bind with high affinity and selectivity to the minor groove of double-stranded DNA. We show how these polyamides can be used as a sequence-specific glue for structural DNA nanotechnology which is orthogonal to Watson-Crick base pairing. For example, we used these polyamides to arranged two double-stranded c-shaped fragments before a ring-closure for the construction of structurally defined double-stranded DNA catenanes.
In the second half of the talk a novel oligonucleotide amplification method will be presented. Synthetic oligonucleotides are the main cost factor for studies in DNA nanotechnology, genetics and synthetic biology that require thousands of these at high quality. Inexpensive chip-synthesized oligonucleotide libraries can contain hundreds of thousands of distinct sequences, however only at sub-femtomole quantities. Here we present an oligonucleotide amplification method based on three rounds of rolling circle amplification. Our method allows the direct production of single-stranded oligonucleotides and is scalable to produce nmole amounts of oligonucleotides per milliliter reaction. In a multistep one-pot procedure, subsets of hundreds or thousands of single-stranded DNAs of different lengths can selectively be amplified and purified together. These oligonucleotides were used to fold several DNA nanostructures. The amplification cost is typically around US$ 20/ nmole oligonucleotides produced (total) and is so far dominated by the use of commercial enzymes.
1 Fraunhofer Institute for Cell Therapy and Immunology, DNA Nanodevices unit, Leipzig, Germany
2 Fakultät für Physik/ Center for Nanoscience, Ludwig Maximilians Universität, Munich, Germany
3 Fraunhofer Institute for Cell Therapy and Immunology Nanotechnology unit, Leipzig, Germany
In the following we present our work towards the development of methods for the organization of single-walled carbon nanotubes (SWNTs) with the help of DNA-based nanostructures. Our goal is a high yield of precise SWNT attachment, placement and orientation, which still keep the intrinsic electrical properties of the SWNT intact or minimize any modification thereof. Previously described schemes for the positioning of SWNT on sheet-like DNA origami templates are based on the dispersion of carbon nanotubes (CNTs) by single stranded DNA (ssDNA) and the subsequent use of rather complex base pairing [1,2,3] or linking via biotin-streptavidin [4] for attachment to the underlying templates.
In a first technique, SWNTs (6,5) dispersed by Polysorbate 20 (Tween 20), are functionalized via electrically non-disruptive π-stacking with bifunctional Pyrene derivatives [5]. In our work we show that these molecules replace Tween 20 and introduce functional groups, like maleimides or succinimidyl esters, without disrupting the electronic or the mechanical properties of the nanotubes. These binding sites allow us to attach proteins to the sidewalls of SWNTs in defined orientations. This will allow direct binding to underlying DNA nanotemplates displaying placement strands containing, e.g., patterns of 3’ biotin modifications, which define the position and alignment of the nanotubes.
Furthermore we present a DNA origami-based modular spacer for DNA-dispersed SWNTs. Here the binding process is based on different affinities of ssDNA sequences toward the sidewalls of the SWNTs. With the help of physiosorption, one can attain precise control over lateral spacing of SWNTs in the nanometer-regime and has the ability to prevent the formation of SWNT bundles in coordination with dielectrophoresis (DEP) methods for bulk alignment. These methods are aiming to build a field–effect transistor with a minimization of restricting percoaltion pathways for electrons.

-
Fig. 1: TEM image of SWNT w/ pyrene maleimide bound to ferritin
Fig. 2: AFM image of DNA-Origami based SWNT spacer bound to individual SWNT
[1] H.T. Maune et al. Nature Nanotechnology 5, 61-66 (2010).
[2] Z. Zhao et al. Org. Biomol. Chem. 11(4), 596-598 (2013).
[3] A. Mangalum, et al. J. Am. Chem. Soc. doi:10.1021/ja312191a (2013).
[4] A. Eskelinen , et al Small, 7(6), 746–750 (2011).
[5] Robert J. Chen, et al. J. Am. Chem. Soc. 2001, 123, 3838-3839
Fraunhofer Institute for Cell Therapy and Immunology (IZI), Perlickstr. 1, 04103 Leipzig, Germany
Currently, the programmed self-assembly of DNA strands via techniques such as "DNA origami" or "DNA bricks" provides the most precise method to construct nanoscale objects of predefined size, shape and composition. Due to their size, biocompatibility and ability to be associated with nearly any type of biomolecule in any desired arrangement (Fig. 1), these objects are promising candidates for molecular therapeutics. To this end, several basic functional necessities are key, including stability in biologically relevant fluids and the ability for molecular cargos to be loaded and released in an efficient manner.
Biostability can be addressed through encapsulation in a protective lipid bilayer, which can shield the internal structure from degradative enzymes and conditions. Here, we used spontaneous self-assembly of a composite lipid bilayer around DNA origami nanostructures, which was directed by electrostatic interactions and triggered by a simple solvent-exchange procedure (Fig. 2) [1]. As analyzed by fluorescence correlation spectroscopy (FCS) and transmission electron microscopy (TEM), a population of encapsulated nanostructures can be assembled.
Previously, naturally DNA-binding molecules such as doxorubicin [2] or siRNA [3] have been successfully transported to cells via DNA-based nanostructures. Taxanes such as paclitaxel (Taxol) are a class of hydrophobic drugs that primarily act upon the microtubule cytoskeleton but also possess DNA-binding capabilities. We use DNA origami-based carriers to transport paclitaxel to cells to target the active, mechanical machinery of cell division and transport. Initial results show paclitaxel-loaded DNA carriers decrease proliferation in breast cancer primary cells and cell lines. Further tests to assess efficacy and effects on microtubule-based processes are underway.
Funding by the Fraunhofer Attract program is gratefully acknowledged.

-
Fig. 1: AFM visualization of streptavidin molecules placed on DNA origami sheetin "IZI" arrangement.(scale bar: 100 nm)
Fig. 2: Schematic (left) and TEM images of lipid-encapsulated DNA-origami 24-helix bundle rod. Adapted from [1]. (scale bar: 100nm
[1] D. M. Smith et al., Nanomedicine 8 (2013), 105-121.
[2] Y. X. Zhao et al., ACS Nano 6 (2012), 8684-8691; Q. Jiang et al., J. Am. Chem. Soc. 134 (2012), 13396-13403.
[3] H. Lee et al., Nat. Nanotech. 7 (2012), 389-393.
1 Leibniz-Institut für Photonische Technologien, Jena, Germany
2 School of Biological, Biomedical and Environmental Sciences, University of Hull, UK
3 Nanotheranostics@CIGMH, DCV, Universidade Nova de Lisboa, Caparica, Portugal
4 Hull York Medical School and Hull and East Yorkshire NHS trust, Hull, UK
5 STABVIDA, Caparica, Portugal 6 microLIQUID, Arrasate – Guipuzcoa, Spain
7 Moltech Srl, Filignano, Italy
The LungCARD project [1] aims to develop a device for a use in personalized medicine in a way that supports the decision making for the appropriate medical treatment of patients with non-small cell lung cancer (NSCLC). The device allows the identification of gene mutations (affecting the expression of Epidermal Growth Factor Receptor - EGFR) in the circulating tumour cancer (CTC) cells obtained from non small cell lung cncer patients’ blood samples. If the specific mutation is identified then receptor tyrosine kinase inhibitors (Gefitinib, erlotinib or afatanib) can be prescribed.
Currently, there are several bottlenecks associated with existing PCR-based diagnostic techniques: i ) use of an invasive and potentially dangerous procedure to obtain complex samples (tumour biopsy embedded in Formalin Fixed Paraffin, FFPE), ii) the extensive expertise of a clinical geneticist is required to accurately interpret the information provided and to setup the best line of therapy and treatment and iii) the assays are quite expensive and time-consuming and can only be carried out at central hospitals with specialized laboratories.
In our approach all the analytical steps necessary for obtaining the information about the EGFR mutations are performed in a single microfluidic chip inserted into a chip reader device. The operator has only to fill up the cartridge with a patient’s blood sample and the results will be obtained within 2-3 hours. The CTC cell isolation from blood is performed using magnetic beads. The cells are then lysed and specific DNA targets are PCR amplified on-chip. The target DNA is hybridized with metal nanoparticle (mNP) labelled probes and separated by capillary electrophoresis. The separation time of the mNP labelled probes with hybridized analyte DNA are optically detected due to the strong and specific absorption of metal NP.
The present work will show the latest development of the chip design, magnetic separation of the CTC cells from blood, engineering of the amplicons, on chip PCR amplification and the optical detection of electrophoretically driven mNP labelled DNA probes.
[1] http://www.lungcard.eu
Department of Chemistry, Graduate School of Science & WPI-iCeMS, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
Direct observation of the movement of biomolecules including enzymes and DNAs should be one of the ultimate goals for investigating the detailed mechanical behavior of the molecules during the reactions. We designed various DNA nanostructures using DNA origami method for the preparation of single-molecule observation scaffolds. Using the designed DNA scaffold and high-speed atomic force microscopy (AFM), the single-molecule behaviors of the DNA modifying enzymes, repair enzymes, and recombinases were observed in the target double-stranded DNAs (dsDNAs) placed in the DNA frame structure. DNA structural changes including G-quadruplex formation and B-Z DNA conformational change were also visualized. Using this system, we observed the photo-induced DNA hybridization and dissociation by detecting the global structural changes of the incorporated two dsDNAs in the DNA frame structure. A pair of azobenzene-modified oligonucleotides (ODNs) was employed, which forms duplex in the trans-form and dissociates in the cis-form. During UV-irradiation, hybridized azobenzene-modified ODNs at the center dissociated, and the subsequent visible-light irradiation induced the hybridization of the photoresponsive ODNs, meaning that the reversed switching behavior such as the hybridization and dissociation was directly visualized at the single-molecule level. These photoresponsive ODNs were also used for controlling assembly and disassembly of the hexagonal DNA origami structures with photoirradiation. The combination of the designed DNA scaffold modified with target DNA strands and high-speed AFM is valuable for visualizing and analyzing the single enzymatic and chemical reactions.
[1] Direct and Single-Molecule Visualization of the Solution-State Structures of G-Hairpin and G-Triplex Intermediates Rajendran, A.; Endo, M.; Hidaka, K.; Sugiyama, H. Angew. Chem. Int. Ed. in press.
[2] Single-Molecule Imaging of Dynamic Motions of Biomolecules in DNA Origami Nanostructures Using High-Speed Atomic Force Microscopy Endo, M.; Sugiyama, H. Acc. Chem. Res. in press.
[3] Dynamic Assembly/Disassembly Processes of Photoresponsive DNA Origami Nanostructures Directly Visualized on a Lipid Membrane Surface Suzuki, Y.; Endo, M.; Yang, Y.; Sugiyama, H. J. Am. Chem. Soc. 2014, 136, 1714-1717.
[4] DNA Origami Based Visualization System for Studying Site-Specific Recombination Events Suzuki, Y.; Endo, M.; Katsuda, Y.;Ou, K.; Hidaka, K.; Sugiyama, H. J. Am. Chem. Soc. 2014, 136, 211-218.
[5] State-of-the Art High Speed Atomic Force Microscopy for the Investigation of Single-Molecular Dynamics of Proteins Rajendran, A.; Endo, M.; Sugiyama, H. Chem. Rev. 2014, 114, 1493-1520.
[6] HIV-1 Nucleocapsid Proteins as Molecular Chaperones for Tetramolecular Antiparallel G-Quadruplex Formation Rajendran, A.; Endo, M.; Hidaka, K.; Tran, P. L. T.; Mergny, J-L.; Gorelick, R. J.; Sugiyama, H. J. Am. Chem. Soc. 2013, 135, 18575-18585.
[7] Direct and Real-time Observation of Rotary Movement of a DNA Nanomechanical Device Rajendran, A.; Endo, M.; Hidaka, K.; Sugiyama, H. J. Am. Chem. Soc. 2013, 135, 1117-1123.
* Kure National College of Technology, 2-2-11 Agaminami, Kure, Hiroshima, 737-8506, JAPAN
** RIKEN, OLABB, Osaka University 6-2-3, Furuedai, Suita, Osaka 565-0874, JAPAN
Organic dyes, fluorescent proteins, and semiconductor quantum dots are common imaging probes in both basic biology and medical diagnostic studies for the real-time monitoring of biochemical process in vitro and in vivo. Besides these, noble-metal (Au, Ag and Pt) nanoclusters have been gaining attention as next-generation bioimaging probes owing to their sub-nanometer size, low cytotoxicity and size-dependent photoluminescent wavelength. In a previous report, we presented the synthesis of blue-emitting Pt5 nanoclusters (em: 470 nm, ex: 380 nm). However, their excitement by UV irradiation risks autofluorescence, phototoxity and strong light scattering. Therefore, we sought nanoclusters with longer wavelength emissions for bioimaging purposes.
In this presentation, we introduced the synthesis of green-emitting Pt nanoclusters (em: 520 nm, ex: 460 nm) by reducing Pt ions from pre-equilibrated Pt/PAMAM (G4-OH) complexes in the dark for 24 h at 4 °C using a mild reductant. Importantly, based on Pt/PAMAM (G4-OH) complex absorbance before reduction, we found our pre-equilibration method could trap more Pt ions in PAMAM (G4-OH) than previous methods. The structural characteristics of the resulting Pt nanoclusters, Pt8L8 (L = C2H2O2S), were determined by electrospray ionization mass spectroscopy. These nanoclusters possess a 28 % quantum yield and are brighter than either Au or Ag nanoclusters. In addition, since Pt nanoclusters have considerably low cytotoxicity, our green-emitting Pt nanoclusters offer great promise for biomedical imaging. Using this method, we expect to extend the photoluminescence wavelength of Pt nanoclusters to the near infrared region, which is often preferable for in vivo imaging experiments.
This work was supported by JSPS KAKENHI Grant Number 25810100.
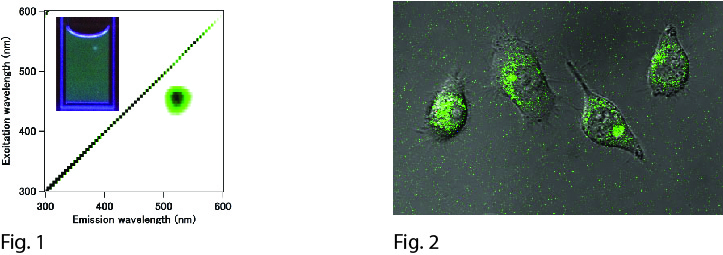
-
Fig. 1: Excitation-emission matrices spectrum of the Pt8 nanoclusters. (Inset: Fluorescent image of the Pt8 nanoclusters in water under UV (365 nm) irradiation.)
Fig. 1: Laser confocal fluorescent microscopic image overlaid with differential interference contrast images of living HeLa cells labeled with Pt8 nanoclusters.
[1] S. Tanaka, Optical Materials Express 3 (2013) 157–165.
[2] S. Tanaka, Angewandte Chemie International Edition 50 (2011) 431-435
Rudolf Virchow Center for Experimental Biomedicine, University of Würzburg, Josef Schneider Str. 2, 97080 Würzburg, Germany
* Department of Chemistry and Pharmacy, Ludwig-Maximilian University Munich, Butenandtstr. 5-13, 81377 Munich, Germany
DNA modifications play essential roles in vivo. Specific structural features in the DNA serve as target sites for protein-DNA interactions, which are responsible for many fundamental control mechanisms of biologically essential processes. Atomic force microscopy (AFM) analyses provide unique and important structural and functional information on such protein-DNA interactions at the level of the individual molecules. Different types of DNA structures can be mimicked by artificially prepared DNA substrates. We have optimized strategies to produce specific motifs in DNA that are frequently employed targets of protein interactions at exactly known positions within long DNA fragments, allowing for a distinction between specific and non-specific protein-DNA interactions in the AFM images and for separate conformational analyses of the different types of protein-DNA complexes [1]. Using this approach, we were able to unravel strategies of the DNA lesion search and recognition mechanism of the Xeroderma pigmentosum group D protein (XPD) of nucleotide excision DNA repair (NER) [2]. NER is responsible for the repair of a broad spectrum of structurally highly diverse types of DNA lesions, most famously of carcinogenic UV light induced photoproducts, such as cyclobutane pyrimidine dimers (CPDs) as well as bulky adducts in genomic DNA. Detailed protein binding position and DNA bend angle analyses on different types of DNA target structures allowed us to show for the first time that XPD preferably recognizes a bulky lesion on the translocated strand, whereas a CPD lesion is preferentially detected on the opposite non-translocated strand.

-
Fig. 1: AFM image of XPD-DNA complexes at lesion site (red arrow) or bound non-specifically (white arrow).
[1] Büchner and Tessmer, Journal of Molecular Recognition 26 (2013) 605-617
[2] Büchner et al., Journal of Biological Chemistry 289 (2014) 3613-3624
Leibniz Institute of Photonic Technology, Albert-Einstein-Straße 9, 07745 Jena, Germany
Since metal nanoparticles (meNP) show a localized surface plasmon resonance (LSPR), they enable an efficient light interaction from far- to near-field and backwards [1]. This gives the opportunity to create or manipulate structures in the nanometer range with light or use the specific resonance-shift as a sensing signal. MeNPs in cooperation with DNA give a further powerful tool to create new materials and functionalities. With the combination of these methods it is possible to fabricate well-defined bottom-up structures like nanoantennas, superlenses and highly sensitive nanobiosensors.
For this issue we present a microfluidic platform to synthesize well defined and highly reproducible silver nanoprisms. Thereby allows the advantages of microfluidics a better control of reaction parameters, which results in a high quality, quantity and reproducibility of silverprisms. In fact, this is needed, since the LSPR-peak position can be fine-tuned over the whole visible spectra and beyond with respect to the prism size (edge-length) (Figure 1) [2].
We will further show the requirement of stabilization of silver nanoprisms by using native ones in DNA-buffer solution. A balanced relation of Au, deposited on silver direct after the synthesis is necessary [3] and will result in a thin Au-layer on the prism (Figure 2) to protect them for any further steps, like the DNA-conjugation. After that, an easy and fast (5 min) direct attachment of DNA on the silver nanoprisms is shown [4].
With respect to edge lengths, different sizes of Au protected prisms can be prepared using these methods. This protection layer allows the preparation of stable DNA-nanoparticle conjugates. Therefore it is possible to generate a toolbox of silver nanoprisms and align them in a highly ordered manner by utilizing DNA-origamis as template substrates (Figure 3).

-
Fig. 1: Overview of silver nanoprisms (solution and droplet) different sizes and corresponding spectral positions
Fig. 2: SEM image of Au enhanced silver prisms
Fig. 3: AFM image of an origami structure with an Au enhanced and DNA labeled silver nanoprism
[1] Csaki, A., et al.,Philosophical transactions. Series A, Mathematical, physical, and engineering sciences, 2011. 369(1950): p. 3483-96.
[2] Aherne, D., et al., Advanced Functional Materials, 2008. 18(14): p. 2005-2016.
[3] Aherne, D., et al., the ACS journal of surfaces and colloids, 2009. 25(17): p. 10165-73.
[4] Zhang, X., M.R. Servos, and J. Liu, Journal of the American Chemical Society, 2012. 134(17): p. 7266-9.
Department of Physics, Nanoscience Center, P.O. Box 35, FI-40740 University of Jyväskylä
* Aalto University School of Science and Technology, P.O.Box 15100, FI-00076 AALTO, Finland
DNA origami as a self-assembled, non-periodic structure has great potential in nanoelectronics and nanophotonics. Previously, we have studied the conductance of individual rectangular origami [1] and TX-tile assemblies by using dielectrophoretic (DEP) trapping and subsequent impedance spectroscopy (IS) as well as DC-current measurements, and found that the conductance of DNA origami is very low in dry, but becomes higher in high humidity [2,3]. Data analysis based on the IS also suggest that the water molecules play a big role in DNA origami conductance, which means the later developed 3D DNA origami may have an improved conductance since their compact inner structure can hold more organized water molecules.
We have trapped and measured 3D DNA origami with the shapes of “brick” and "L". The results proved that the individual 3D DNA origami can also well be manipulated and trapped by DEP. Against the expectations, the conductance measurements have so far shown no evidence about any significant improvement in conductance. However, some localized but dramatic destruction of the silica substrate after trapping have been observed, the detailed cause of this are still under investigation.
We have also utilized a DNA-structure consisting of two TX-tiles (A and B) forming a definite chain of three tiles, i.e. BAB, as a scaffold for gold nanoparticles (AuNPs) functionalization: Each tile has an AuNP attached in the middle so that the fully assembled structure consists of a sequence of three AuNPs which form the needed metallic islands for the aimed single electron transistor (SET). The fabricated BAB-AuNP conjugates were trapped by DEP and the IV-curves were measured from room temperature to 4.2 K. To obtain high enough tunneling current, the AuNPs of the conjugated and trapped structure were further grown by electrochemical Au-deposition (Nanoprobes – cat. # 2113). After that the IV measurements revealed clear Coulomb Blockade even at room temperature on couple of samples.
Funding by the Academy of Finland (project numbers 218182, 263868) and Emil Aaltonen foundation (Post Doc –pool) are gratefully acknowledged.
[1] P. W. K. Rothemund, Nature 440 (2006) 297-302.
[2] V. Linko et al., Small 5, 2382–2386 (2009).
[3] V. Linko et al., Nanotechnology 22, 275610 (2011).
Institute of Cellular and Molecular Biology, Medical School and Chemical Nanoscience Laboratory, School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
As the computing capacity of devices is increasing and the size of electrical gadgets are decreasing, nano-scale systems are required to allow further progress if this trend is to continue. DNA conducting wires have the capability of tolerating this development.
The ability to use DNA as a template to engineer a conducting wire has been a desirable concept for several years. However, the production of such a conducting strand has been a major challenge in nanotechnology advancements. The intrinsic conductivity of DNA is not adequate for these applications, therefore, modifications to normal nucleotides, such as 6-thio-deoxyguanosine triphosphate (-GTP), are applied to allow chelation of metals – producing a conducting wire.
A PCR-based method has been developed, which is capable of annealing the duplexes to form ‘sticky ends’, followed by extension with an Archael Pyrococcus furiosus Family B Polymerase variant, Z3 (Fig. 1). This method produced DNA duplexes of up to 50,000 base pairs in length (approximately 17µm) after only 20 PCR cycles. This length is adequate for the use as a conducting wire. The sequences included poly[A].poly[T], poly[G].poly[T], [AG]/[TC], [AAG]/[TTC], [AAAG]/[TTTC] and [AAAAG]/[TTTTC] showing a large variety can be used.
To use this elongated DNA for a conducting nano-scale function, a development in the method to incorporate modified bases is required. Z3 has shown a potential to efficiently incorporate modified bases, leading to a speculation that this method is promising for the production of conducting wires. A variety of modified nucleotides are now available which expands the possible nanotechnology applications possible via this method of elongation.

-
Fig. 1: Heat-cool cycles of ds-DNA. The double strand is heated to separate the strands and cooled quickly so the most thermostable position cannot be formed. This allows mis-alignment when re-annealing the strands. The DNA polymerase, Z3, binds new dNTPs, producing an extended template.
Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel
The information encoded in the base sequence of DNA provides substantial structural and functional information. These features of nucleic acids allow the use of DNA as a material for novel applications. These will be addressed by describing the implementation of DNA in the following areas:
- The development of DNA machines, and particularly, interlocked DNA ring structures (catenanes and rotaxanes). The synthesis of a five-ring catenane (olympiadane) and a seven-ring catenane will be introduced.
- The organization and reconfiguration of Au NPs structures by means of the DNA machines, and the control of switchable plasmonic phenomena in the structures will be described.
- The development of switchable DNA hydrogels undergoing hydrogel-solution or hydrogel-solid transitions will be exemplified. Particularly, shape-memory DNA hydrogels will be introduced.
- The development of “smart” DNA-functionalized mesoporous SiO2 nanoparticles and stable DNA capsules will be introduced, and the use of the systems as drug carriers for controlled release, will be discussed
1 Institute of Photonic Technology, Albert-Einstein-Straße 9, 07745 Jena, Germany
2 Nanoscience Center, Department of Physics, P.O. Box 35, FI-40014 University of Jyväskylä, Finland
The interaction between intense laser pulses and metal nanoparticles offers the contact-free nanoscale manipulation of biological molecules immediately adjacent to the excited nanoparticles. In our studies, we´re using the nonlinear excitation of metal nanoparticles by their treatment with fs-laser pulses for inducing an excitation transfer along DNA molecules. The excitation transfer was firstly demonstrated by the micrometer-wide destruction of an electron-sensitive polymer layer, following the original position of the former DNA molecules (Fig.1). In a further experimental approach we used fluorescence labeled silver nanoparticles (AgNP´s), interconnected by DNA molecules in a distance of a few micrometers. Here, the induced excitation transfer by the laser excitation resulted in the bleaching of the acceptor-AgNP´s (Fig.2). Furthermore, we examined the behavior with more stable gold nanoparticles (AuNP´s), where the excitation transfer seems to be dependent from the polarization angle of the incident laser light, leading to DNA destruction (Fig.3).
This novel excitation transfer phenomenon in molecular wires could have implications as interconnects and functional devices in molecular nanofabrication and electronics [2].
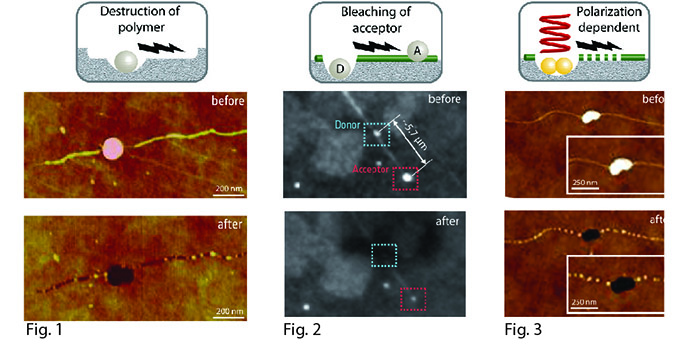
-
Fig. 1: AFM images of AgNP-labeled DNA molecules before and after their treatment with fs laser pulses.
Fig. 2: Fluorescence images of AgNP-labeled DNA molecules before and after their treatment with fs laser pulses.
Fig. 3: AFM images of AuNP-labeled DNA molecules before and after their treatment with fs laser pulses.
[1] Wirth, J., et al., Plasmonic Nanofabrication by Long-Range Excitation Transfer via DNA Nanowire. Nano Letters, 2011. 11(4): p. 1505-1511.
[2] Toppari, J.J., et al., Plasmonic Coupling and Long-Range Transfer of an Excitation along a DNA Nanowire. Acs Nano, 2013. 7(2): p. 1291-1298.
[3] Wirth, J., et al., Plasmonically-enhanced electron escape from gold nanoparticles and their polarization-dependent excitation transfer along DNA nanowires. submitted
Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah, USA
The fabrication of inorganic nanostructures with controlled shape, composition and dimensions is critical for nanodevice manufacturing. We are creating a wide variety of scaffolded DNA origami [1] nanostructures that have thin (~10 nm) linewidths, multiple branching points, asymmetric junctions, and overall dimensions of a few hundreds of nanometers [2]. We have focused on the selective deposition of inorganic materials onto these DNA templates, through the usage of silver and palladium seeding methods. Subsequent electroless plating of metals including gold, palladium and copper results in nanostructures that have features as narrow as 30 nm and the same shape as the initial DNA structure, as shown in Figure 1 [3-4]. We also utilize site-specific localization of gold nanoparticles within DNA origami to seed the plating of metal nanowires in selected places and to create controlled nanogaps, as depicted in Figure 2 [5]. We have characterized these metallized DNA structures by atomic force microscopy and electron microscopy, and have also studied their electrical properties (Figure 3) [5,6]. Our conductive DNA nanowires have strong potential for utilization in nanofabrication, electronics, sensing and photonics.
This work was funded in part by the Semiconductor Research Foundation under contract # 2013-RJ-2487.

-
Fig. 1: AFM image of gold metallized T-shape DNA origami; scale bar is 200 nm.
Fig. 2: Logic gate prototype. (left) DNA origami design; (right) SEM image of the plated structure.
Fig. 3: (a) I-V plot of an electrode pair bridged by two DNA nanowires. (b) Histogram of 12 measured resistance values.
[1] Rothemund, P. W. K. Nature, 440, 297-302 (2006).
[2] Pound, E.; Ashton, J. R.; Becerril, H. A.; Woolley, A. T. Nano Lett., 9, 4302-4305 (2009).
[3] Liu, J. F.; Geng, Y. L.; Pound, E.; Gyawali, S.; Ashton, J. R.; Hickey, J.; Woolley, A. T.; Harb, J. N. ACS Nano, 5, 2240-2247 (2011).
[4] Geng, Y.; Liu, J.; Pound, E.; Gyawali, S.; Harb, J. N.; Woolley, A. T. J. Mater. Chem., 21, 12126-12131 (2011).
[5] Pearson, A.C.; Liu, J.; Pound, E.; Uprety, B.; Woolley, A.T.; Davis, R.C.; Harb, J.N. J. Phys. Chem. B 116, 10551-10560 (2012).
[6] Geng, Y.; Pearson, A.C.; Gates, E.P.; Uprety, B.; Davis, R.C.; Harb, J.N.; Woolley, A.T. Langmuir 29, 3482-3490 (2013).